
In a previous blog post, I talked about how the hbond action command in CPPTRAJ is capable of outputting a file that shows whether or not a hydrogen bond formed across a series of time steps as indicated by a 1 or a 0 (for clarification, click on this link: http://williamkennerly.com/blog/helpful-cpptraj-commands-part-2-action-commands/). I recently found out that this is not entirely true. When I first used CPPTRAJ’s hbond command, the data I obtained only showed 0’s and 1’s in the solute-solvent hydrogen bond column. At the time, I had made the assumption that these 0’s and 1’s simply indicated whether or not hydrogen bonds formed at a certain time step, 0 meaning no hydrogen bonds were formed and 1 meaning one or more hydrogen bonds were formed. The reason my data yielded strange results was because of two things: a default angle cutoff for bond formation set at 135 degrees, and a default distance cutoff set at 3Å. These cutoffs meant that if a bond was to form at an angle smaller than 135 degrees or at a distance greater than 3Å, then CPPTRAJ does not count it and eliminates the data for that bond entirely.
After changing the cutoffs (I eliminated the angle cutoff by specifying the angle to be -1, and changed the distance cutoff to be 3.5Å), I obtained very different data. The following is the exact syntax that I used in my .traj file to get my output:
parm mlt_wat.prmtop
trajin mdcrd/prod.mdcrd
hbond trpHBond :19 out trp.HBvTime2.dat angle -1 dist 3.5 image solventdonor :WAT solventacceptor :WAT@O solvout trp.HBAvg2.dat
run
quit
This .traj file uses a .mdcrd file that contains the final result of a production run of a whole Melittin molecule; however, by specifying the residue number (:19) I only extracted information about the hydrogen bonds formed with the respective tryptophan atoms that form them (the two Nitrogens and the Oxygen), which when plotted results in the graph seen at the bottom of this post. The problem with this obtained data is that it is most likely not reliable. While looking at the average angles of the hydrogen bonds, it was discovered that the angles were very acute ranging from 62° to 106°. They also had slightly longer bond lengths then the original cutoff of 3Å, the lengths ranging from 3 to 3.2Å. Therefore, it can be assumed that these bonds are unlikely to happen in nature.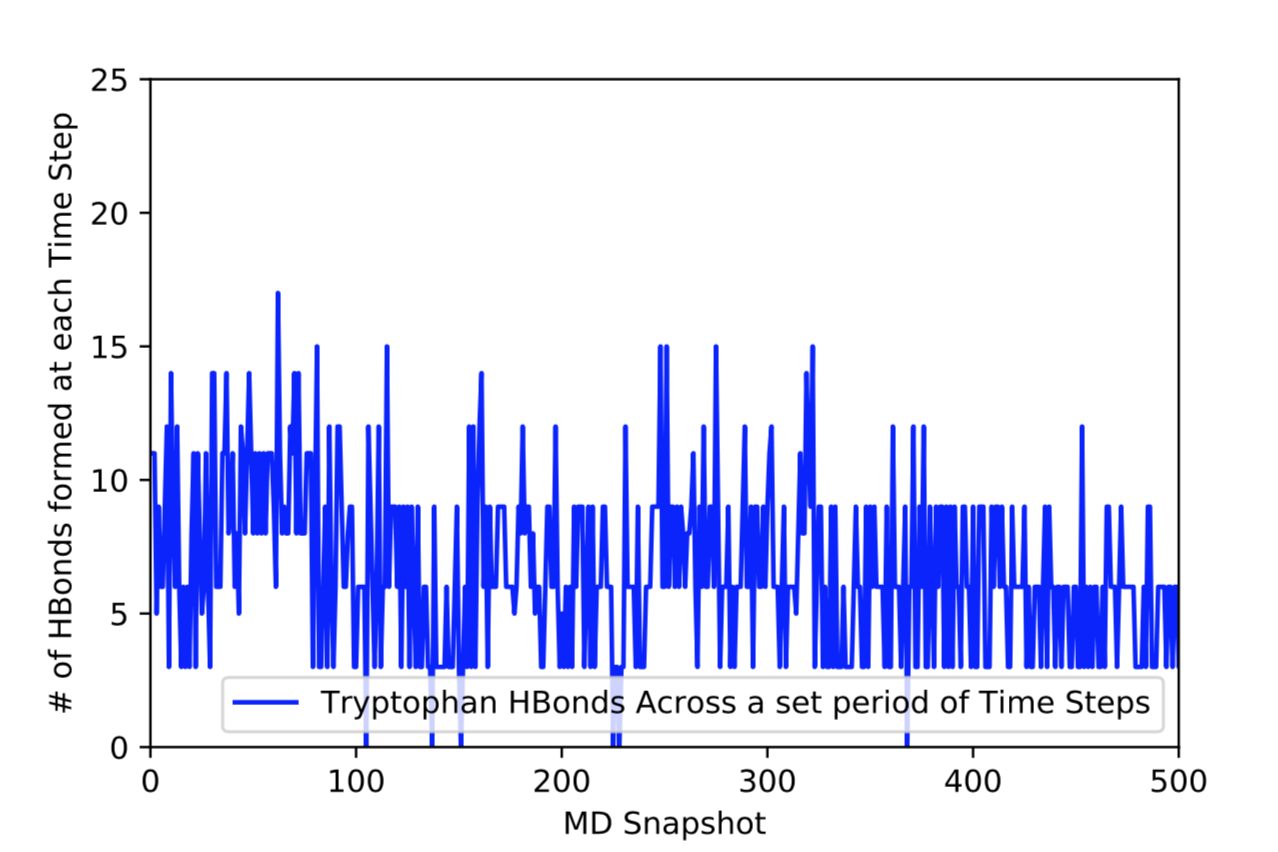
Hi Will – when you mention a previous blog post, it would help to link to it for clarity.
I can definitely see why relaxing the cutoffs would increase the number of hydrogen bonds counted. How ‘bad’ are these ‘new’ hydrogen bonds — do they have very acute angles, or close to the original 135 cutoff?
Lastly, I could swear you had changed one more thing about these hbond applications. Didn’t you also in the prior run only look at the Trp in the mellitin , and for the data mentioned in this post look at all solute-solvent h-bond contacts? Correct me if I’m wrong. To clarify for your post, it would be helpful to mention what system and trajectory you are analyzing and the specific hbond command syntax.