
CPPTRAJ has a variety of commands for analyzing MD Simulation outputs, but because there are so many commands it would be very difficult to describe them all in detail in a single blogpost. As such, this first post will consist of the descriptions of three Topology Commands that I found to be useful for my research of the Tryptophan residue in Melittin. These Topology Commands print out Molecular Topology related information in CPPTRAJ’s interactive mode. For all intents and purposes, the visuals used in this blogpost will strictly be regarding Melittin.
Note: [<mask>] indicates a single residue/atom or range of residues/atoms
atominfo [<mask>]
This command prints out general information on each atom specified by the given [<mask>] modifier. The information is outputted into 12 columns. The following visual is an example output of the command being called on the 19th residue of a Melittin .mdcrd file.

- #Atom refers to the atom’s index as given by Amber16
- Name refers to the atom’s name identifier as given by Amber16
- #Res refers to the residue number in which the atom is located
- 2nd Name refers to the shorthand name of the residue in which the atom is located
- #Mol refers to the atom’s molecule number
- Type refers to the type of atom in the residue (i.e. alpha, beta, etc)
- Charge refers to the electron charge of the atom
- Mass refers to the mass of the atom
- GBradius refers to the generalized Born radius of the atom
- E1 refers to the element symbol
- rVDW refers to the Van der Waal’s force radius of the atom
- eVDW refers to the epsilon Van der Waal’s force of the atom
Example Usage: atominfo :19
bondinfo [<mask>]
This command prints out general information in the form of 6 columns about each bond between each atom as specified by the [<mask>] modifier. The following visual is an example output of the command being called on a specific carbon atom in the 19th residue of a Melittin .mdcrd file.

- Bond refers to the bond index as specified by Amber16
- Kb refers to the bond force constant
- Req refers to the bond equilibrium value in Angstroms
- atom names refers to the names of the bonded atoms as specified by Amber16
- (numbers) refers to the atom indexes as specified by Amber16 as well as the types of atoms that are bonded together
Example Usage: bondinfo :19@CA
resinfo [<mask>]
This command prints out general information in 7 columns about a single residue or range of residues as specified by the given [<mask>] modifier. The following visual is an example output of this command being called on the range of residues that make up Melittin. The reason I had to indicate all the residues was because the .mdcrd file that I used had Melittin simulated in a water box. If the command was called without specifying the [<mask>] modifier, the output will include the residue info for all the water molecules in the water box as well. This goes for the atominfo and bondinfo commands too.
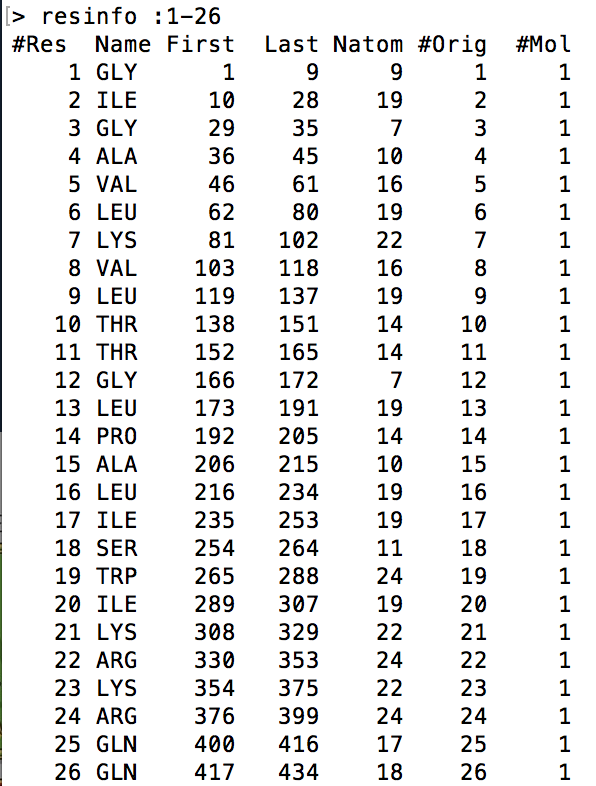
- #Res refers to the residue index as specified by Amber16
- Name refers to the shorthand name for each residue
- First refers to the atom index of the first atom in the residue
- Last refers to the atom index of the last atom in the residue
- Natom refers to the number of atoms in the residue
- #Orig refers to the original residue number in the original PDB file of which the date comes from
- #Mol refers to the molecule number
One can also add the modifier, [short], to display the residues in the FASTA code sequence form. For Melittin, the sequence would look like this: GIGAVLKVLTTGLPALISWIKRKRQQ.
Example Usage: resinfo :1-26