This past weekend, our research group attended talks by guest speakers, presented our abstracts, and fielded questions by our posters at the 2016 MERCURY Conference at Bucknell University.
There were six guest speakers: Chris Cramer, Steven Wheeler, Jeffrey Evanseck, Chris Wilmer, Richard Pastor, and Kate Holloway.
Cramer gave a crash course in quantum mechanics and computational chemistry. He referred to the psi operator in the Schrödinger equation as an oracle, and he said it is important to ask the oracle something you know before you ask it something you don’t know, i.e. to validate a computational method before using it to predict new information. He stated that the total energy of a molecule can be determined exclusively from its electron density. However, he pointed out self-interaction error as the biggest error in DFT, in which the calculation averages the position of one electron, so that in the calculation, the election repels itself (impossible). This error needs to be corrected for in the calculation. After the crash course, he discussed his research with MOFs (Metal Organic Frameworks), porous materials on the nano scale that can store and separate liquids or gases.
Wheeler delineated important goals and advice for computational chemists. He proposed that you only really learn how quantum chemistry methods work by programming them yourself, that you should learn a programming language (especially Python), and that you should take a course in linear algebra, if possible. Wheeler described three main types of functionals: semi-empirical (approximate/Hartree-Fock theory for 1000s of atoms), ab initio (MO-based methods for 10s to 100s of atoms, depending on the level of theory), and DFT (for 20 to 200 atoms). He asserted that choosing the appropriate level of theory for calculations is a primary focus of computational chemists. He mentioned his trials to find an appropriate functional with DFT for his research. Because traditional DFT functionals completely fail to capture dispersion interactions, he decided on B97-D, as a fast, accurate functional, though it had problems with long-range interactions. His research group studies the pi-pi* stacking of substituted benzene rings.
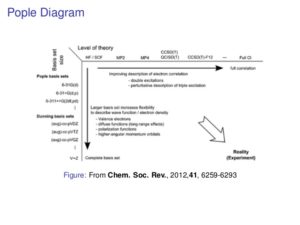
Evanseck introduced molecular dynamics. He stated three reasons for computation: to interpret experimental data, to extend (fill gaps) in experimental data, and to make predictions to guide the experiment. He made an analogy to the Pople Diagram (see below), with force field sophistication instead of level of theory. He then broke down the pros and cons on molecular dynamics methods vs. quantum methods. In addition, he pointed out that it is much easier to tweak the existing potential energy functions than to write new ones.
Wilmer has become successful with a start-up called NuMat Technologies Inc., which develops new MOFs with programs that general hypothetical MOFs and calculates their ability to store and separate natural gases for motor vehicles and CO2 capture. He advanced with the help of some business majors by participating in business plan competitions.
Pastor informed us of the tremendous difficulty and computing time of membrane simulations, specifically looking at natural antibiotics in epithelial layers. He said it was appropriate for early grad students to be inexperienced and lazy (to learn but not disrupt the work of the professors or pursue fruitless projects), post-docs to be smart and hard-working (to effectively collaborate with professors), and PIs to be smart and lazy (to delegate tasks properly and be able to respond to post-docs and students). He encouraged students to seek truth, work within a community, and to be a good and generous friend by keeping friends out of the wrong quadrants (smart/inexperienced, hardworking/lazy), where inexperienced and hardworking was the most dangerous for everyone.
Holloway has worked on finding cures for AIDS and hepatitis C at Merck. She described her career trajectory, having decided on computational chemistry because she was good at it and didn’t like lab work. She became a computational chemist at Merck straight from her Ph.D. She emphasized the cost (>$2.5 billion) and waiting time (>10 years) for new drugs to get on the market. However, potentially saving people’s lives motivated her and her fellow researchers.
Presenting abstracts seemed like it would be easy enough, but I got some of the ordering of my spiel wrong and flubbed some of the wording. This experience has taught me that memorizing and practicing a short speech might be a good idea in the future.
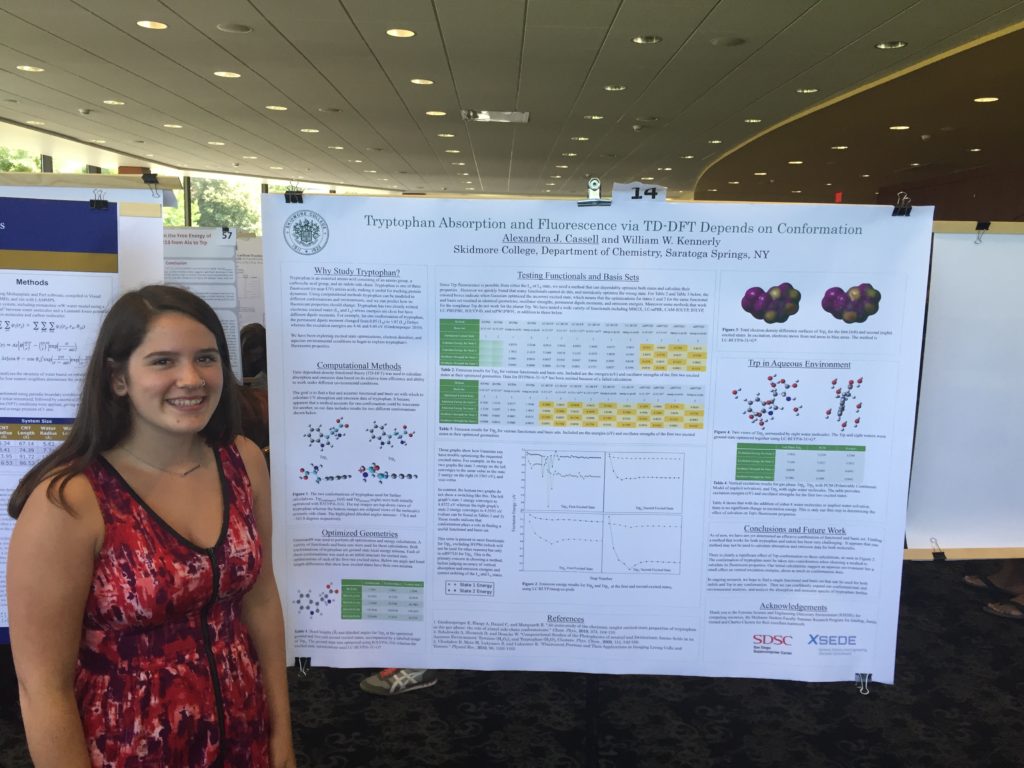
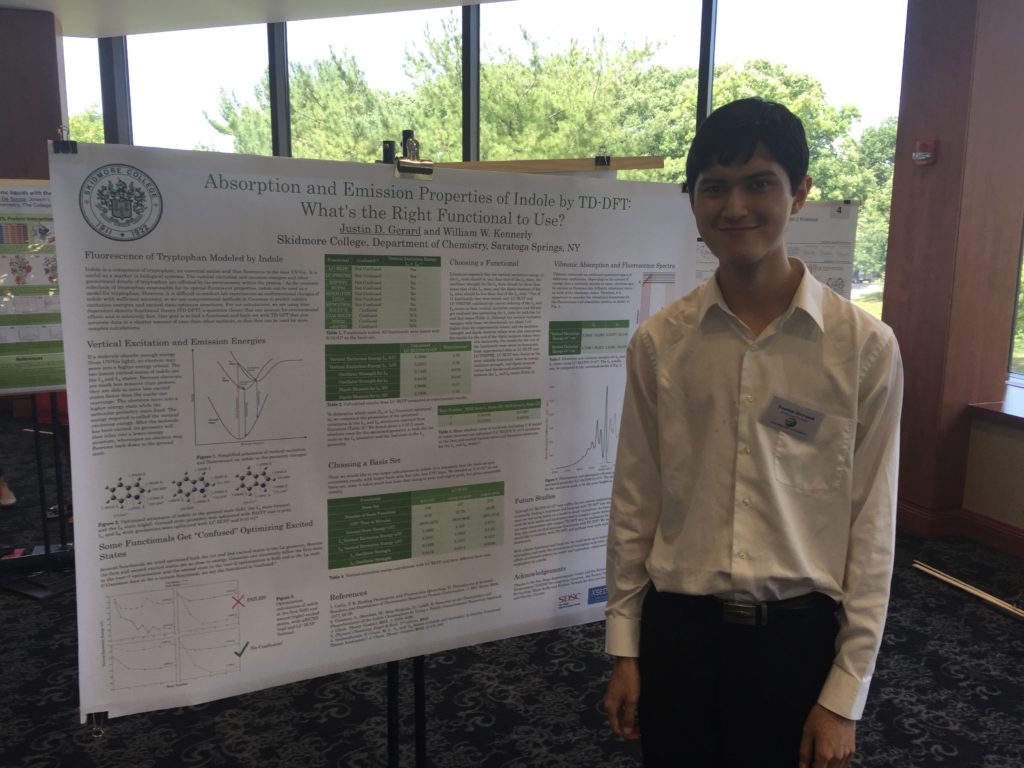
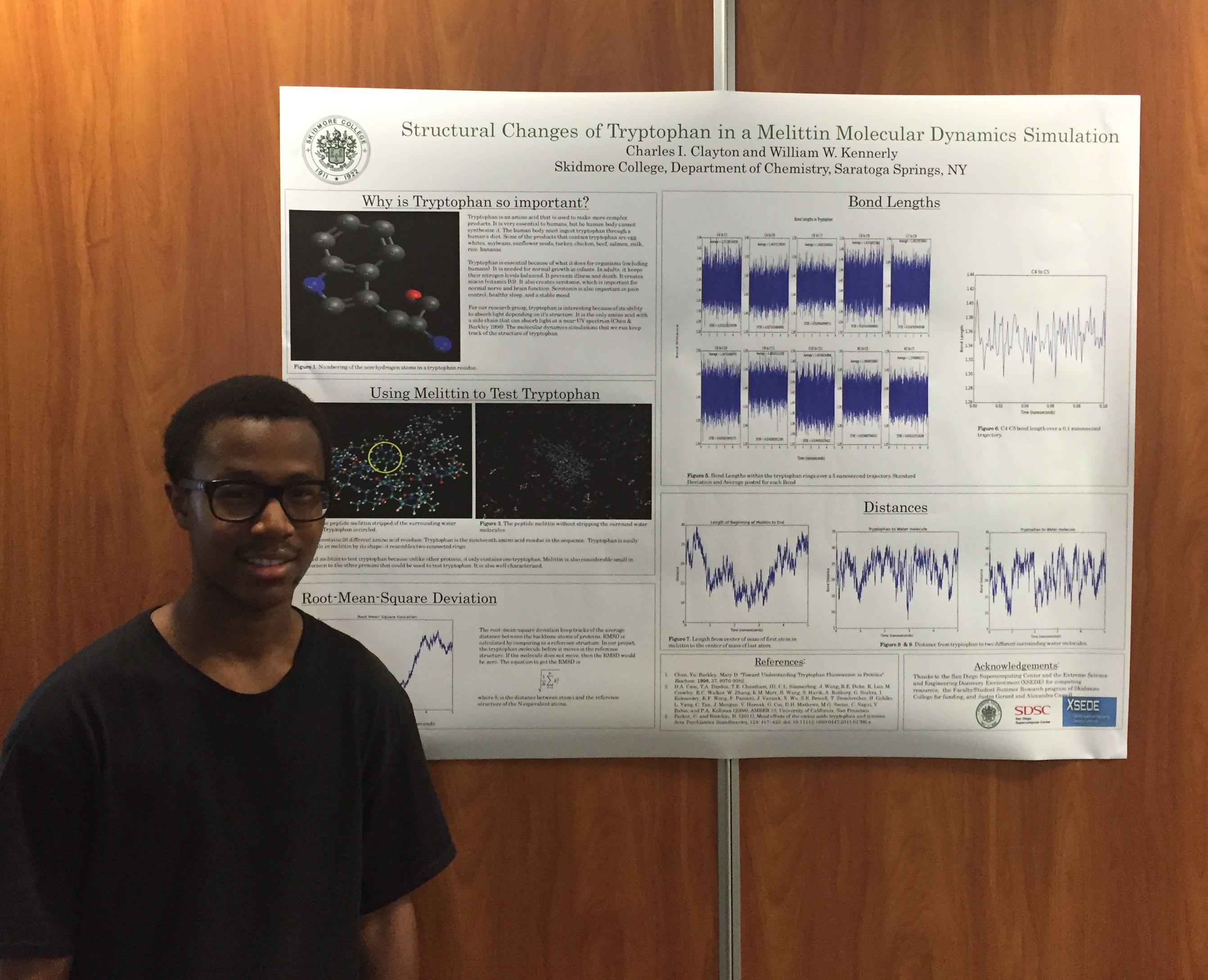
Poster presentations went much better for me. Because we had rehearsed, I was comfortable with the flow of my presentation, and I adjusted the length and detail of it based on the knowledge of the person listening (anyone from Steven Wheeler to students with no knowledge of Gaussian). One student named Clorice was particularly interested in my poster, because she was also starting some excited state optimizations with TD-DFT. However, she did not experience the same difficulty optimizing first and second excited energy states, because her project involved pi-pi* stacking and enzyme activity, not a molecule with fluorescent properties similar to indole.
A highlight of the stay was hearing Chris Cramer expertly sing some folk songs at karaoke night (not to mention the our own group’s hoe-down of “Sweet Home Alabama”). That night, along with conversations with students and faculty in chemistry who are also involved in music, gave me hope that music and art will stay relevant to me if I pursue a career in chemistry.